의료기기는 유럽 연합으로의 판매를 위하여 유럽 의료기기 규정에 따라 적합함을 나타내는 CE 마크를 부착해야 합니다.
인증개요
새롭게 개정된 유럽 의료기기 규정인 Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 (이하 MDR)은 유럽 연합 내에서 판매 되는, 인간을 대상으로 하는 의료기기 및 해당 의료기기의 액세서리를 규제하는 규격입니다.
MDR은 기존에 적용되던 의료기기 지침인 Council Directive 93/42/EEC on Medical Devices (이하MDD) 및 전기 전자 이식형 의료기기 지침인 Council Directive 90/385/EEC on Active Implantable Medical Devices (AIMDD)를 대체하게 됩니다.
MDR은 2020년 5월 26일부터 완전히 시행 될 계획이었으나, 전 세계적인 코로나 바이러스의 유행으로 인하여 적용날짜가 1년간 연기되어 2021년 5월 26일부터 완전히 적용 되었습니다.
더하여, 체외 진단 의료기기(Invitrodiagnostic medical devices)의 새로운 규정인 Regulation (EU) 2017/746 (IVDR)은 다가오는 2022년 5월부터 적용되며, 기존의 체외 진단 의료기기지침인 Directive 98/79/EC (IVDD)를 대체하게 됩니다.
의료기기 등급 분류
의료기기의 등급은 기기의 의도된 목적과 내재된 위험을 고려하여 MDR의 부속서 VIII 등급 분류 규칙에 따라 진행됩니다.
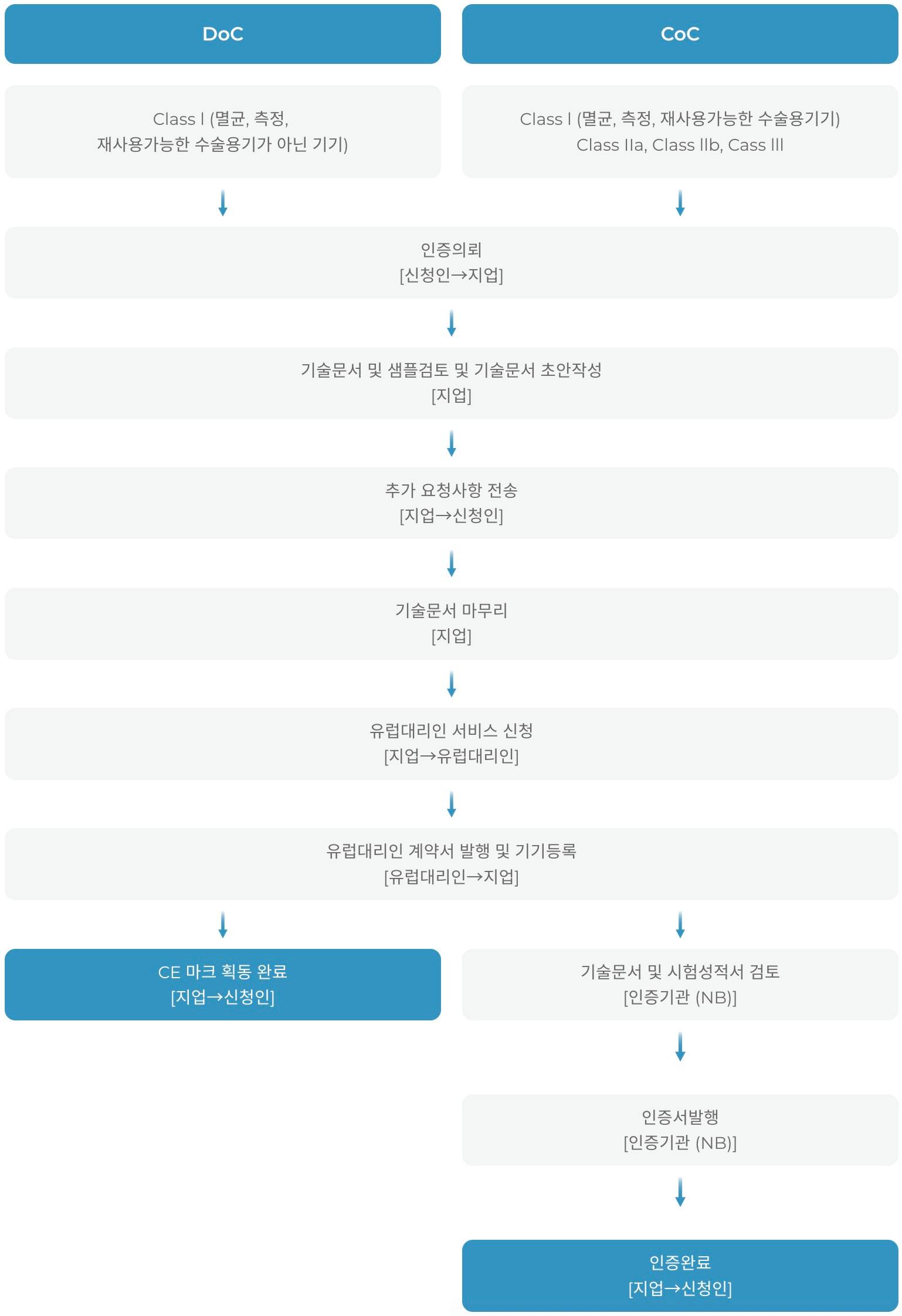
의료기기의 등급별 해당하는 적합성 평가 절차에 따라 요구사항의 준수가 입증 된 경우, 의료기기 제조자는 제 19조에 따라 EU 적합성선언(EU Declaration of Conformity, EU DoC)을 작성하고 제 20조에 따라 CE 마크를 부착해야 합니다.
(주문제작 또는 임상조사용 기기는 별도)
하기 적합성 평가 절차는 의료기기의 특성에 따라 다르게 적용될 수 있습니다.
I 등급
Annex II → Annex III → Annex IV → Annex V
IIa 등급
a) Chapters I and III of Annex IX → Section 4 of Annex IX → Annex IV → Annex V
b) Part A of Annex XI → Annex II → Annex III → Annex IV → Annex V
c) Part B of Annex XI → Annex II → Annex III → Annex IV → Annex V
III 등급
a) Chapters I and III of Annex IX → Section 4 of Annex IX → Annex IV → Annex V
b) Annex X → Part A of Annex XI → Annex IV → Annex V
c) Annex X → Part B of Annex XI → Annex IV → Annex V